Successful Strategies for the Analysis of EtG and EtS in Urine
Rugged Sample Preparation and Analysis Conditions for High-Throughput Labs

Introduction
Ethyl glucuronide (EtG) and ethyl sulfate (EtS) are important biomarkers for monitoring alcohol use. Detecting these metabolites has proven beneficial for abstinence enforcement and zero tolerance treatment programs requiring assessment of alcohol consumption. The analysis of EtG and EtS in urine offers many advantages for abstinence monitoring, including a three-day detection window, good stability in properly stored specimens, and analytical specificity. However, EtG and EtS are both polar compounds, making them difficult to retain via reversed-phase chromatography (Figure 1). Both compounds are also very sensitive to matrix interferences, so accurate results can be difficult to obtain if retention is not sufficient to resolve EtG and EtS from the sample matrix. Further complicating this analysis is the inherently variable nature of human urine from person to person and population to population, let alone differing health states, and a myriad of other sources of variation. If matrix suppression occurs, low limits of detection may not be met. Even more important, if coeluting matrix components are isobaric to the compounds of interest, then accurate quantitation may be impossible.
Typical LC-MS/MS methods for EtG/EtS analysis have several shortfalls: poor retention and resolution of EtG and EtS from matrix components, long run times that limit sample throughput, and short column lifetimes. These common issues are all related to the separation characteristics of the chromatographic system, as well as to some sample preparation considerations and mass spectrometer settings. This study describes several strategies to address these issues; however, it is difficult for a single analytical solution to be suitable for the numerous urine sample types submitted for EtG/EtS testing. Therefore, this work also explores ways to improve method sensitivity for lower detection limits and provides helpful techniques for mitigating troublesome matrix effects.
Figure 1: Structures of EtG and EtS.
The Fundamentals
One of the principal problems with the analysis of EtG and EtS in urine is separating the target compounds from interfering matrix components, which is particularly difficult due to the lack of retention typically observed using reversed-phase columns and conditions. Coeluting matrix components can affect the ionization of the analytes of interest, even if the matrix components are not monitored in the MRM channels. And, for this application, there are matrix components that commonly appear in the monitored MRM channels, making coelution a considerable problem for identification and quantitation.
Building a Foundation Method
To reliably separate polar EtG and EtS analytes from confounding matrix interferences, you must start with retention. Only by retaining the compounds of interest will you be able to make the appropriate method development choices that will then separate them from the matrix. The Raptor EtG/EtS column is designed specifically for this application. Its novel stationary phase offers multimode retention characteristics that effectively and consistently separate the target analytes from matrix interferences. For this reason, it was chosen for the development of a base method for reliable analysis of EtG and EtS in urine.
As an initial step, we conditioned a new column to ensure consistent retention times, adequate separation from matrix peaks, and good peak shapes. We strongly recommend conditioning the analytical column prior to use with 30 injections of 20x dilute-and-shoot matrix samples that run through the full gradient program. For this base method, the matrix samples were prepared by aliquoting 50 µL of ethanol-free human urine and diluting it 20x with 950 µL of a working internal standard (25 ng/mL EtS-d5 and 100 ng/mL EtG-d5 in 0.1% formic acid in water). Diluted samples were then vortexed, centrifuged, and injected on a Raptor EtG/EtS column for column conditioning prior to sample analysis.
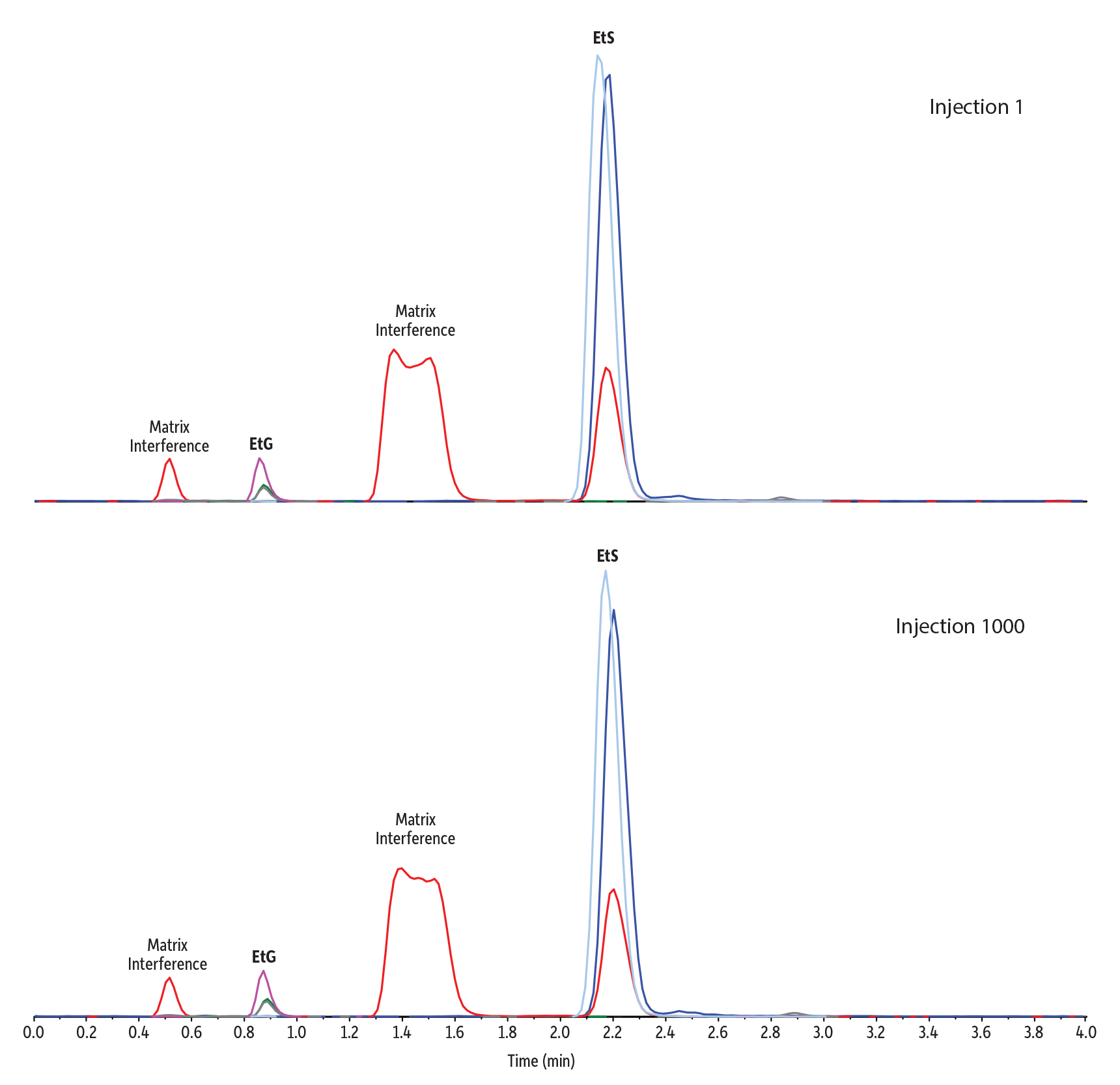
Following column conditioning, this simple 20x dilute-and-shoot sample preparation was coupled with LC-MS/MS conditions that ensured retention and effectively separated EtG and EtS from potentially interfering matrix components in a fast, 4-minute analysis (Figure 2). No increased system pressure or changes in peak shape or retention time were observed—even after 1000 urine matrix injections—indicating no column clogging had occurred. The conditions proved to be highly reliable and long-lasting, which will benefit high-throughput laboratories by minimizing downtime for column changes.
Figure 2: Rugged Raptor EtG/EtS columns provide separation of EtG and EtS from matrix interferences, even after 1000 injections of matrix.
| Peaks | Precursor Ion | Product Ion | Product Ion | |
|---|---|---|---|---|
| 1. | Ethyl-β-D-glucuronide-d5 | 225.9 | 84.9 | - |
| 2. | Ethyl-β-D-glucuronide | 220.8 | 84.9 | 74.8 |
| 3. | Ethyl sulfate-d5 | 129.7 | 97.7 | - |
| 4. | Ethyl sulfate | 124.7 | 96.8 | 79.7 |
| Column | Raptor EtG/EtS (cat.# 9325A12) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 100 mm x 2.1 mm ID | ||||||||||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||||||||||
| Pore Size: | 90 Å | ||||||||||||||||||||
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 µm frit (cat.# 25809) | ||||||||||||||||||||
| Temp.: | 35 °C | ||||||||||||||||||||
| Standard/Sample | |||||||||||||||||||||
| Diluent: | 0.1% Formic acid in water | ||||||||||||||||||||
| Conc.: | A 500 ng/mL QC sample was prepared in urine. 50 µL of the sample was diluted with 950 µL of a working internal standard (25 ng/mL EtS-d5/100 ng/mL EtG-d5 in 0.1% formic acid in water). The sample was vortexed at 3500 rpm for 10 seconds to mix. The sample was then centrifuged at 3000 rpm for 5 minutes at 10 °C. The autosampler needle was adjusted to inject from the supernatant. | ||||||||||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||||||||||
| Mobile Phase | |||||||||||||||||||||
| A: | 0.1% Formic acid in water | ||||||||||||||||||||
| B: | 0.1% Formic acid in acetonitrile | ||||||||||||||||||||
|
| Detector | MS/MS |
|---|---|
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | HPLC |
| Notes | Reference Standards Ethyl-β-D-glucuronide (cat.# 34101) Ethyl-β-D-glucuronide-d5 (cat.# 34102) Ethyl sulfate sodium salt (cat.# 34103) Ethyl sulfate-d5 sodium salt (cat.# 34104) |
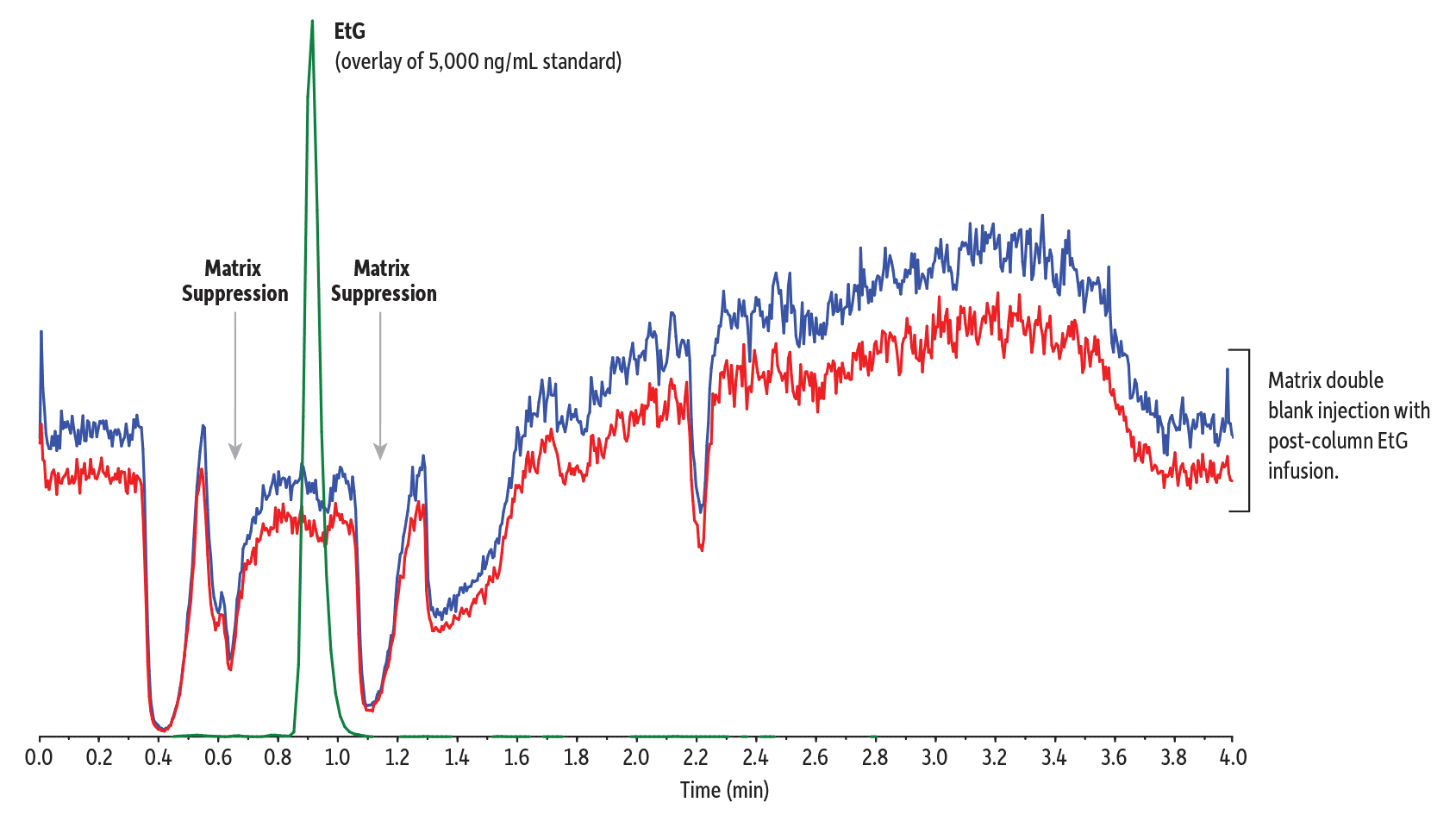
Figure 2 clearly shows that this method effectively separates EtS from isobaric matrix components. It also separates the target analytes from those regions where matrix components are eluting but are not observed in the chromatogram because they don’t share the selected ion transitions of EtG or EtS. In these cases, the presence of the matrix isn’t observed directly, but it can impact the ionization of the target analytes via competition (i.e., matrix effect). This can be visualized by injecting the matrix while scanning a continuous post-column infusion of the analytes of interest. In this case, we focused on EtG because of its much lower response compared to EtS.
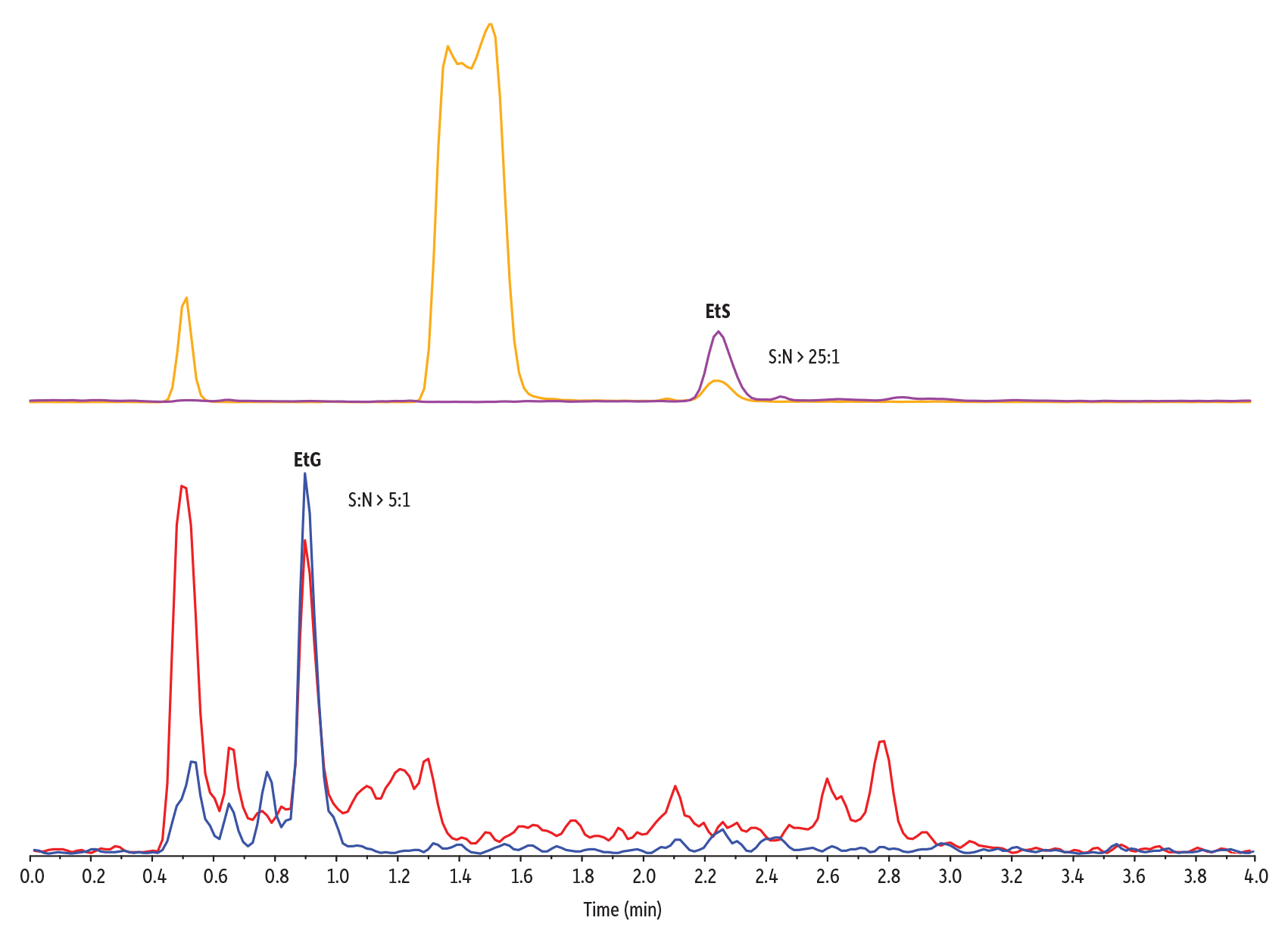
Figure 3 illustrates this effect: dips in the baseline correspond to when matrix components suppress the signal for EtG, which lowers sensitivity and introduces run-to-run irreproducibility. Figure 3 also shows how, under the given method conditions, EtG and EtS elute outside of these ionization suppression regions, which improves sensitivity. Samples prepared in matrix showed approximately 80% (EtG) and 100% (EtS) of the signal obtained when samples were prepared in solvent across four QC levels (50, 150, 750, and 4000 ng/mL), demonstrating that matrix effect is minimal in these samples. Figure 4 demonstrates the low levels of quantitation achievable when matrix compounds do not interfere with the detection of EtG and EtS.
In addition, quantitative method calibration studies showed good linearity with r2 values of 0.999 or greater using linear 1/x weighted regression across a calibration range of 50–5000 ng/mL. Method accuracy and precision results, performed on three different days, were excellent for all QC levels for both EtG and EtS. Table I summarizes the accuracy and precision data for QC samples prepared using the same dilute-and-shoot method outlined above and analyzed using the method shown in Figures 2-4.
Figure 3: EtG elutes outside the matrix suppression zones (double blank injection with post-column EtG infusion).
| Peaks | Precursor Ion | Product Ion | Product Ion | |
|---|---|---|---|---|
| 1. | Ethyl-β-D-glucuronide | 220.8 | 84.9 | 74.8 |
| Column | Raptor EtG/EtS (cat.# 9325A12) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 100 mm x 2.1 mm ID | ||||||||||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||||||||||
| Pore Size: | 90 Å | ||||||||||||||||||||
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 µm frit (cat.# 25809) | ||||||||||||||||||||
| Temp.: | 35 °C | ||||||||||||||||||||
| Standard/Sample | |||||||||||||||||||||
| Conc.: | 50 µL of urine was diluted with 950 µL of 0.1% formic acid in water. The sample was vortexed at 3500 rpm for 10 seconds to mix. The sample was then centrifuged at 3000 rpm for 5 minutes at 10 °C. The autosampler needle was adjusted to inject from the supernatant. A post-column infusion of 1000 ng/mL EtG was made. | ||||||||||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||||||||||
| Mobile Phase | |||||||||||||||||||||
| A: | 0.1% Formic acid in water | ||||||||||||||||||||
| B: | 0.1% Formic acid in acetonitrile | ||||||||||||||||||||
|
| Detector | MS/MS |
|---|---|
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | HPLC |
| Notes |
Figure 4: Low levels of quantitation are met for the analysis of EtG and EtS in urine using this LC-MS/MS method because matrix interferences are avoided (50 ng/mL LLOQ sample).
| Peaks | Precursor Ion | Product Ion | Product Ion | |
|---|---|---|---|---|
| 1. | Ethyl-β-D-glucuronide | 220.8 | 84.9 | 74.8 |
| 2. | Ethyl sulfate | 124.7 | 96.8 | 79.7 |
| Column | Raptor EtG/EtS (cat.# 9325A12) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 100 mm x 2.1 mm ID | ||||||||||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||||||||||
| Pore Size: | 90 Å | ||||||||||||||||||||
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 µm frit (cat.# 25809) | ||||||||||||||||||||
| Temp.: | 35 °C | ||||||||||||||||||||
| Standard/Sample | |||||||||||||||||||||
| Diluent: | 0.1% Formic acid in water | ||||||||||||||||||||
| Conc.: | A 50 ng/mL standard was prepared in urine. 50 µL of the standard was diluted with 950 µL of a working internal standard (25 ng/mL EtS-d5/100 ng/mL EtG-d5 in 0.1% formic acid in water). The sample was vortexed at 3500 rpm for 10 seconds to mix. The sample was then centrifuged at 3000 rpm for 5 minutes at 10 °C. The autosampler needle was adjusted to inject from the supernatant. | ||||||||||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||||||||||
| Mobile Phase | |||||||||||||||||||||
| A: | 0.1% Formic acid in water | ||||||||||||||||||||
| B: | 0.1% Formic acid in acetonitrile | ||||||||||||||||||||
|
| Detector | MS/MS |
|---|---|
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | HPLC |
| Notes | Reference Standards Ethyl-β-D-glucuronide (cat.# 34101) Ethyl-β-D-glucuronide-d5 (cat.# 34102) Ethyl sulfate sodium salt (cat.# 34103) Ethyl sulfate-d5 sodium salt (cat.# 34104) |
Table I: Accuracy and Precision of QC Samples
| QC LLOQ (50 ng/mL) |
QC Low (150 ng/mL) |
QC Mid (750 ng/mL) |
QC High (4,000 ng/mL) |
|||||||||
| Analyte | Average Conc. (ng/mL) | Average Accuracy (%) | %RSD | Average Conc. (ng/mL) | Average Accuracy (%) | %RSD | Average Conc. (ng/mL) | Average Accuracy (%) | %RSD | Average Conc. (ng/mL) | Average Accuracy (%) |
%RSD |
| EtG | 51.2 | 102 | 6.82 | 143 | 95.2 | 4.99 | 749 | 99.8 | 3.68 | 3,949 | 98.7 | 2.04 |
| EtS | 46.9 | 93.7 | 4.01 | 143 | 95.6 | 1.42 | 762 | 102 | 1.00 | 3,958 | 98.9 | 1.59 |
Finally, the robustness of this foundational method was tested by monitoring five patient samples with positive results for EtG and EtS in the lower end of the linear range across multiple instrument platforms on nine manufacturing lots of Raptor EtG/EtS columns. The results presented below in Table II illustrate the method robustness on this column.
Table II: Inter-run precision of patient samples across multiple instrument platforms, multiple days, and multiple column lots (n=9).
| Patient Sample |
Average EtG Concentration (ng/mL) |
%RSD | Average EtS Concentration (ng/mL) |
%RSD |
|---|---|---|---|---|
| A | 216 | 6.13 | 78.0 | 3.56 |
| B | 1,167 | 4.81 | 300 | 3.24 |
| C | 98.2 | 9.76 | 82.4 | 4.01 |
| D | 319 | 8.33 | 233 | 3.63 |
| E | 247 | 11.2 | 163 | 8.70 |
Sensitivity Improvement Strategies
Using the base method established above, we demonstrated that the Raptor EtG/EtS column reliably meets typical validation criteria for the analysis of EtG and EtS in human urine with a lower limit of quantitation of 50 ng/mL. However, there may be applications that require lower detection limits or labs using legacy instruments (e.g., SCIEX API 4000) that may not have the sensitivity of newer models. In these cases, what strategies can help boost sensitivity?
Optimization of MS Ion Source
It’s important to recognize that optimized ionization source settings for highly organic mobile phases may not be optimal for the ionization of compounds that elute under high aqueous mobile phase conditions, as is the case with the analysis of EtG and EtS in urine.
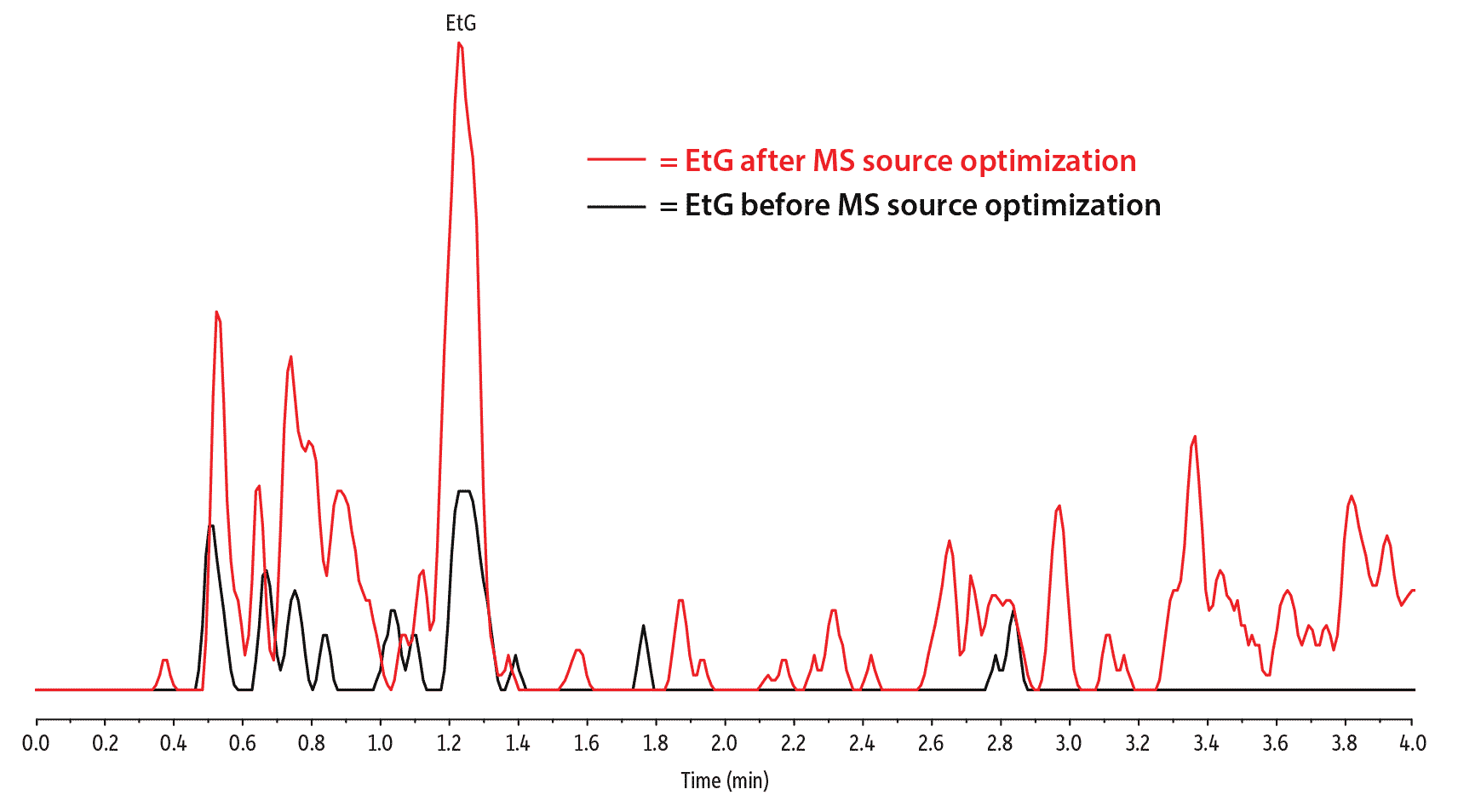
While optimizing MS source parameters is something that will likely require experimental adjustments, higher source temperatures and a readjusted probe position may help provide incremental sensitivity boosts for any LC-MS/MS. In addition to method parameter adjustments, simply cleaning the instrument’s ionization source can provide increased sensitivity. Figure 5 is an illustration of EtG sensitivity before and after source optimization and cleaning.
Figure 5: Cleaning the source and optimizing ionization parameters is an effective way to improve EtG sensitivity.
| Peaks | tR (min) | Conc. (ng/mL) | Precursor Ion | Product Ion | |
|---|---|---|---|---|---|
| 1. | Ethyl-β-D-glucuronide (EtG) | 1.23 | 100 | 221.2 | 85.1 |
| Column | Raptor EtG/EtS (cat.# 9325A12) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 100 mm x 2.1 mm ID | ||||||||||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||||||||||
| Pore Size: | 90 Å | ||||||||||||||||||||
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 μm frit (cat.# 25809) | ||||||||||||||||||||
| Temp.: | 35 °C | ||||||||||||||||||||
| Standard/Sample | Ethyl- | ||||||||||||||||||||
| Diluent: | 0.01% Formic acid in water | ||||||||||||||||||||
| Conc.: | 100 ng/mL (40x acetonitrile precipitation sample cleanup) | ||||||||||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||||||||||
| Mobile Phase | |||||||||||||||||||||
| A: | 0.01% Formic acid in water | ||||||||||||||||||||
| B: | 0.1% Formic acid in acetonitrile | ||||||||||||||||||||
|
| Detector | MS/MS |
|---|---|
| Ion Source: | Electrospray |
| Ion Mode: | ESI- |
| Instrument | HPLC |
| Sample Preparation | Acetonitrile Precipitation: A 100 ng/mL standard was prepared in urine. A 50 μL aliquot of the standard solution was mixed with 10 μL of internal standard (20 μg/mL EtG-d5 and 5 μg/mL EtS-d5) and 150 µL of acetonitrile by vortexing at 3000 rpm for 10 seconds, followed by centrifugation at 4300 rpm for 10 minutes at 10 °C. After centrifugation, 100 µL of the supernatant was diluted with 900 µL (40x dilution) of 0.01% formic acid in water. The sample was then vortexed at 3000 rpm for 10 seconds and injected. |
Mobile Phase Additive Concentration
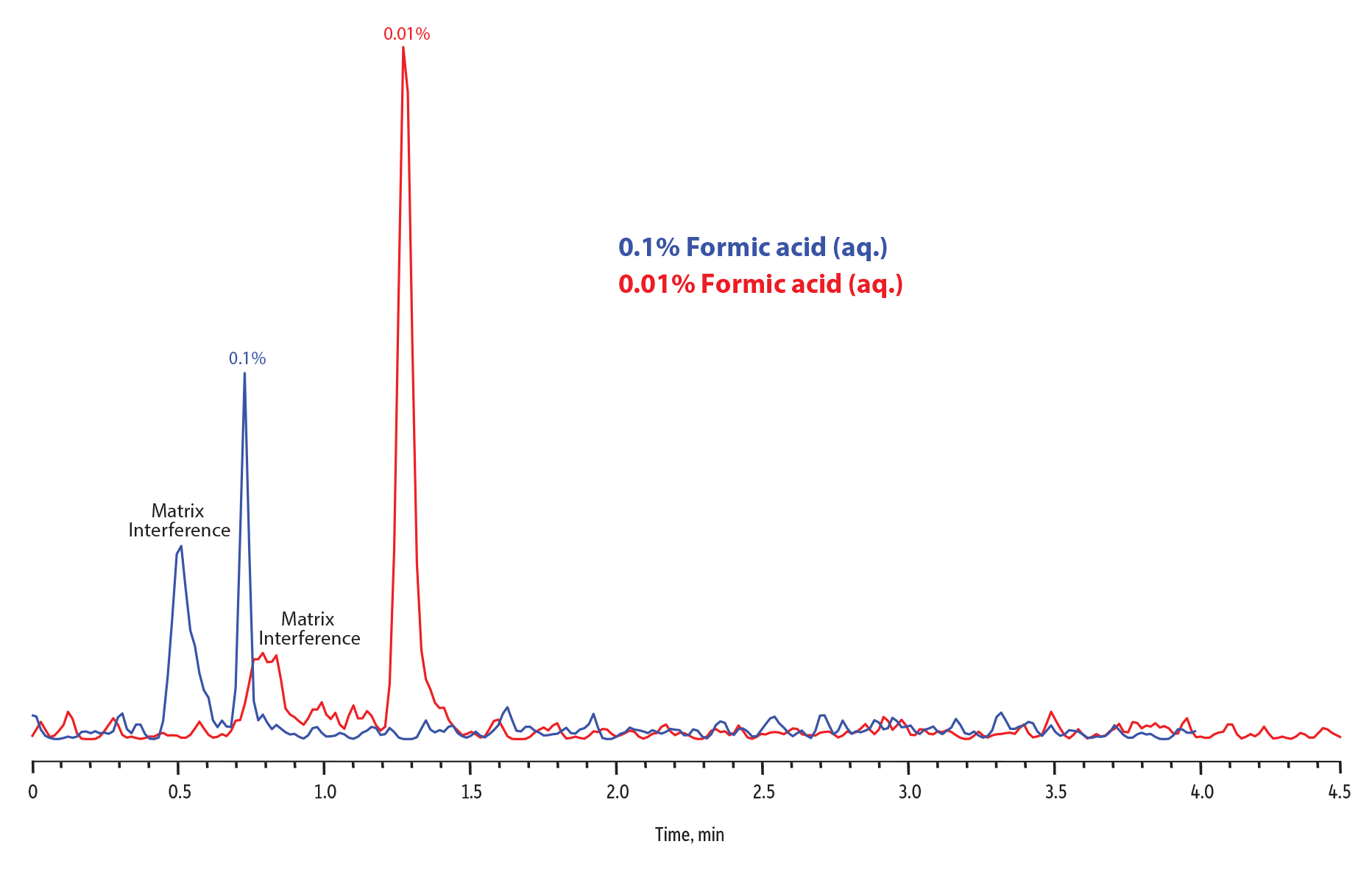
Another technique for improving the sensitivity of EtG in particular is to reduce the concentration of the formic acid in the aqueous mobile phase. Starting with the basic method described earlier in the Fundamentals section, reducing the formic acid concentration in the aqueous mobile phase from 0.1% to 0.01% resulted in an increase in EtG sensitivity. A couple of factors contributed to this effect. First, decreasing the formic acid concentration in the aqueous mobile phase increases EtG retention, and this may lead to further separation from matrix interferences for particularly difficult samples. Second, higher concentrations of formic acid in the mobile phase actually suppress the signal, so reducing the concentration increases sensitivity.
Keep in mind that changing formic acid composition in the aqueous mobile phase can have an effect on overall chromatography. In this case, decreasing the formic acid concentration in aqueous mobile phase increased sensitivity for EtG (Figure 6), but in doing so it also increased the retention of both EtG and EtS, causing EtS to elute during the final ramp of the gradient when the percent aqueous mobile phase concentration was increasing. However, the high concentration of formic acid is critical to the chromatographic performance of EtS on the Raptor EtG/EtS column, and eluting under aqueous mobile phase conditions, when the formic acid concentration was an order of magnitude lower than the original method resulted in severe peak splitting. To combat peak distortion, we simply extended the 5-35% organic mobile phase ramp from 2.5 to 3.0 minutes, allowing EtS to elute under the same mobile phase conditions as in the original method. There is almost always compromise in method development, and in this case, we had to slightly increase analysis time by 0.5 minutes to boost EtG sensitivity while still allowing for great EtS peak shape and adequate column re-equilibration.
In addition, in order to maintain good peak shapes, the sample diluent should be adjusted to match the mobile phase. For this example, blank urine samples were fortified at 25 ng/mL and diluted 20x with either 0.1% formic acid or 0.01% formic acid in water. Samples were then injected and analyzed using aqueous mobile phases with the same formic acid concentrations as the respective sample diluents. Figure 6 illustrates the overall sensitivity gain achieved by decreasing formic acid concentration in the aqueous mobile phase while making the necessary adjustments to the gradient and sample diluent. This strategy provides an excellent option for labs interested in low-level analysis of EtG and EtS in urine.
Figure 6: Improvement in EtG response by dropping the aqueous mobile phase concentration of formic acid from 0.1% to 0.01%.
| Column | Raptor EtG/EtS (cat.# 9325A12) |
|---|---|
| Dimensions: | 100 mm x 2.1 mm ID |
| Particle Size: | 2.7 µm |
| Pore Size: | 90 Å |
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 µm frit (cat.# 25809) |
| Temp.: | 35 °C |
| Standard/Sample | Ethyl- |
| Conc.: | 25 ng/mL |
| Inj. Vol.: | 10 µL |
| Mobile Phase | |
| Flow: | 0.5 mL/min |
| Detector | MS/MS |
|---|---|
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | HPLC |
| Sample Preparation | A 25 ng/mL standard was prepared in MS Gold urine. 50 µL aliquots of the standard were diluted with 950 µL of working internal standard (50 ng/mL EtS-d5/200 ng/mL EtG-d5 in either 0.1% formic acid or 0.01% formic acid in water to match the respective aqueous mobile phases). The samples were vortexed at 3500 rpm for 10 seconds to mix. The samples were then centrifuged at 3000 rpm for 5 minutes at 10 °C and analyzed by LC-MS/MS using their respective mobile phase conditions to compare EtG signal sensitivity between the methods. |
| Notes | Method Comparison Details 0.1% Aqueous Formic Acid Method (Blue) • Sample diluent: 0.1% formic acid in water • Mobile phase A: 0.1% formic acid in water • Mobile phase B: 0.1% formic acid in acetonitrile • Gradient (%B): 0.00 min (5%), 2.50 min (35%), 2.51 min (5%), 4.00 (5%) 0.01% Aqueous Formic Acid Method (Red) • Sample diluent: 0.01% formic acid in water • Mobile phase A: 0.01% formic acid in water • Mobile phase B: 0.1% formic acid in acetonitrile • Gradient (%B): 0.00 min (5%), 3.00 min (35%), 3.01 min (5%), 4.50 (5%) |
Strategies for Dealing with Extreme Matrix Interferences
Even though the basic method described above in the Fundamentals section was easily verified and demonstrated to work on patient samples, some analysts may run into difficult samples where the profile of matrix interferences is different and might coelute with EtG or EtS, which negatively impacts quantitation. In some instances, the proper application of the fundamentals will still allow for good separation of EtG and EtS from matrix interference.
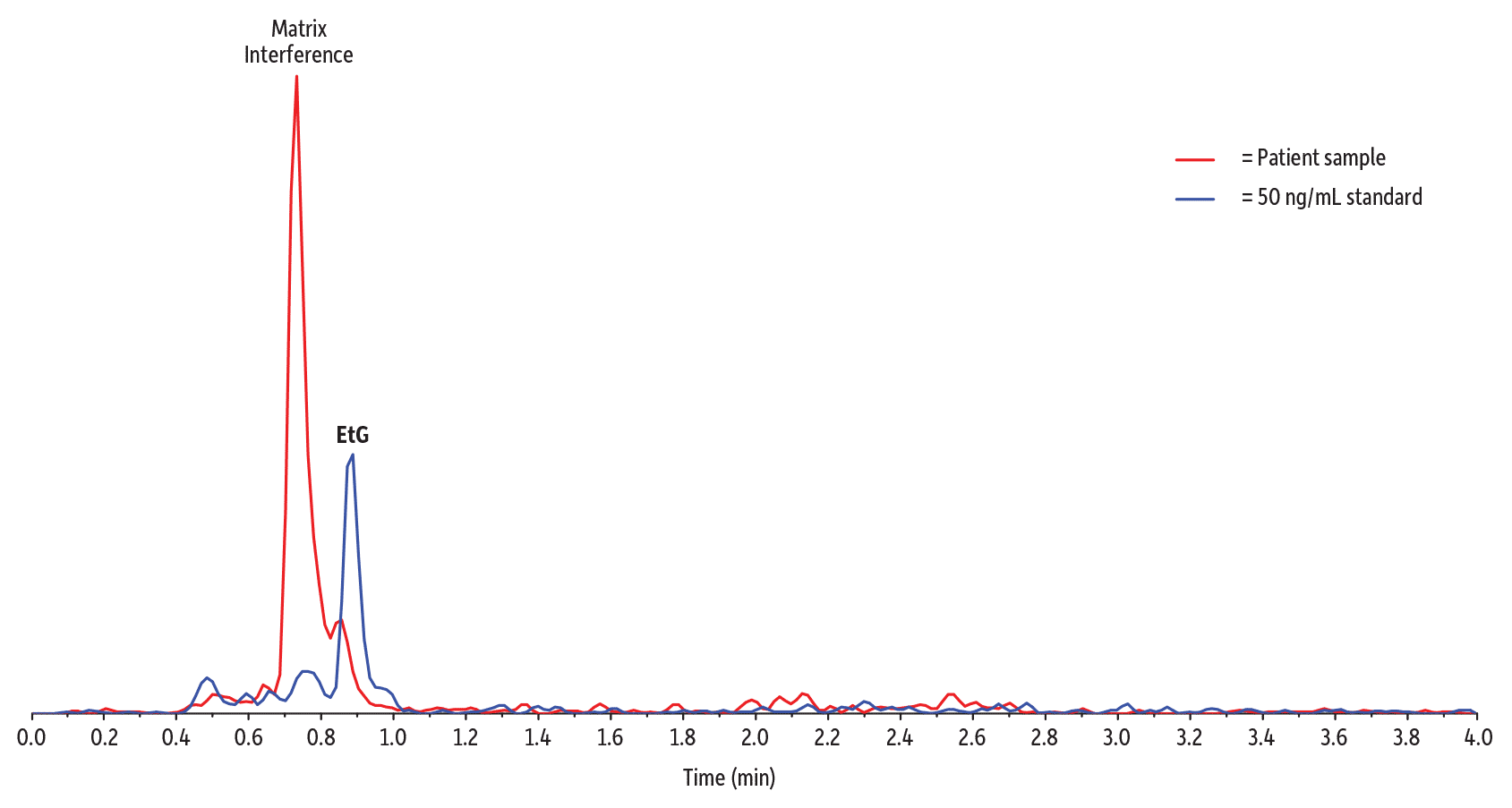
Figure 7 is an example of the foundation method separating EtG from a closely eluting matrix peak that shared the same ion transition as the analyte in a very distinct urine sample, even at the LLOQ concentrations.
Figure 7: Even closely eluting matrix interference is well resolved by the application of a column with adequate retention and a method tuned for separation from matrix interferences.
| Peaks | Precursor Ion | Product Ion | |
|---|---|---|---|
| 1. | Ethyl-β-D-glucuronide | 220.8 | 74.8 |
| Column | Raptor EtG/EtS (cat.# 9325A12) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 100 mm x 2.1 mm ID | ||||||||||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||||||||||
| Pore Size: | 90 Å | ||||||||||||||||||||
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 µm frit (cat.# 25809) | ||||||||||||||||||||
| Temp.: | 35 °C | ||||||||||||||||||||
| Standard/Sample | |||||||||||||||||||||
| Diluent: | 0.1% Formic acid in water | ||||||||||||||||||||
| Conc.: | 50 µL of the sample or standard was diluted with 950 µL of a working internal standard (25 ng/mL EtS-d5/100 ng/mL EtG-d5 in 0.1% formic acid in water). The sample was vortexed at 3500 rpm for 10 seconds to mix. The sample was then centrifuged at 3000 rpm for 5 minutes at 10 °C. The autosampler needle was adjusted to inject from the supernatant. | ||||||||||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||||||||||
| Mobile Phase | |||||||||||||||||||||
| A: | 0.1% Formic acid in water | ||||||||||||||||||||
| B: | 0.1% Formic acid in acetonitrile | ||||||||||||||||||||
|
| Detector | MS/MS |
|---|---|
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | HPLC |
| Notes | Reference Standards Ethyl-β-D-glucuronide (cat.# 34101) Ethyl-β-D-glucuronide-d5 (cat.# 34102) Ethyl sulfate sodium salt (cat.# 34103) Ethyl sulfate-d5 sodium salt (cat.# 34104) |
However, there are some urine samples with much more pronounced matrix components that can interfere with the analysis of EtG and EtS in urine, even when using a fit-for-purpose solution. And, in these cases, the best recourse may be to increase the degree of sample preparation in order to remove the particularly problematic matrix components. The good news is that the additional sample preparation doesn’t have to be any more intense than a simple protein precipitation followed by dilution. In most cases, protein precipitation will take care of the stubborn matrix compounds that can interfere with EtG and EtS.
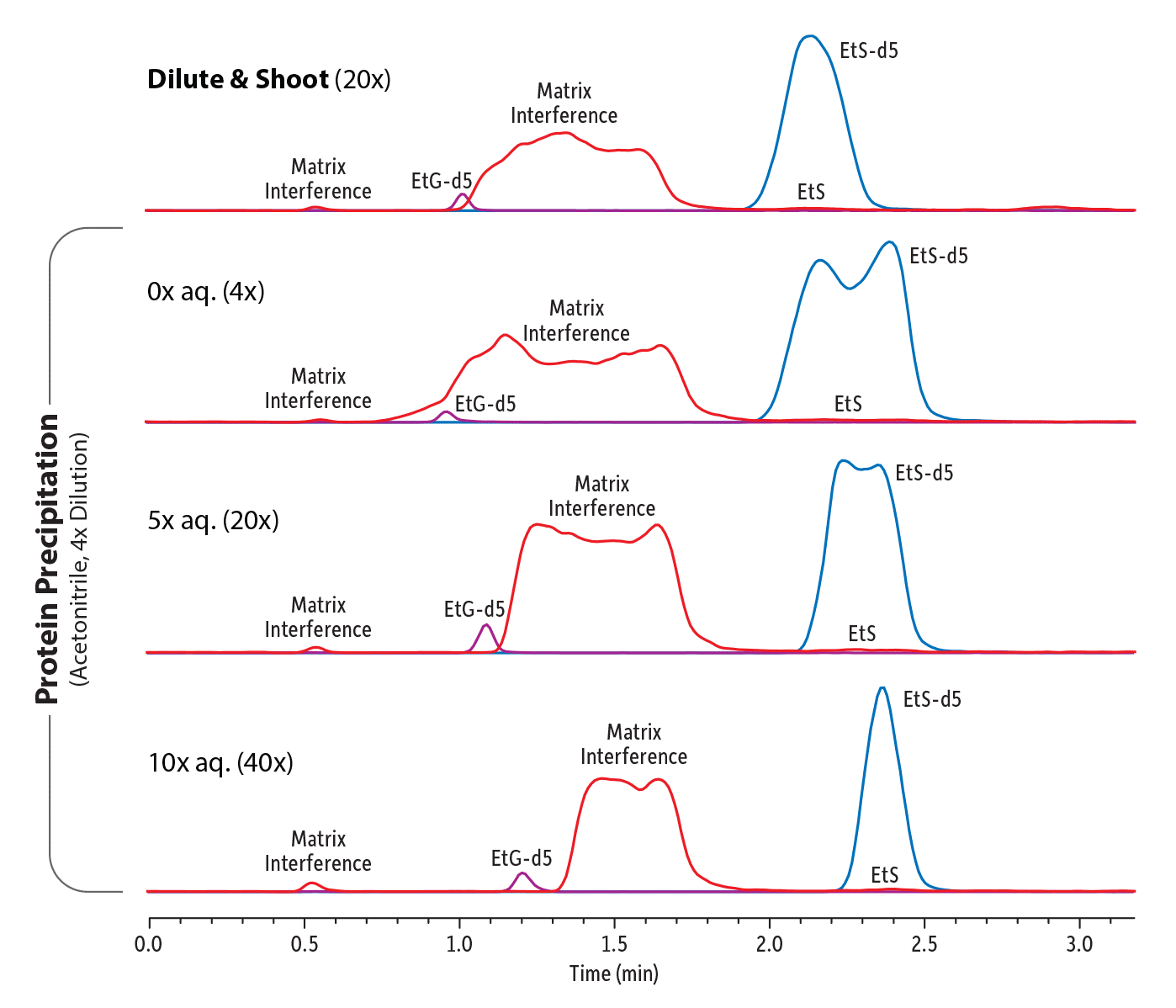
Diluting with water is a very important step because, after protein precipitation, the sample will contain a much higher concentration of organic solvent than a normal dilute-and-shoot sample, and the difference between the sample diluent and the initial LC mobile phase conditions can cause a solvent mismatch, resulting in poor peak shapes for the target analytes. This effect can be so severe that the benefits of protein precipitation are nullified because peak shapes are so distorted that there is still interference between the remaining matrix compounds and EtG and EtS. Sample diluent/mobile phase mismatch can be easily remedied by diluting the sample after protein precipitation with acidified water, making the sample composition more similar to the mobile phase. In our study, diluting the sample 40 times resulted in excellent peak shapes.
Figure 8 illustrates this effect: the top trace is an example of a complex urine sample with particularly intense matrix effects; however, as shown in the second trace, simply performing a protein precipitation and then analyzing the sample without any aqueous dilution did not resolve the issue. The sample diluent/mobile phase mismatch was so extreme that all the peak shapes were severely distorted. However, increasing the aqueous component for a total dilution factor of 20x, and then ultimately 40x, using water that has the same formic acid concentration as the mobile phase (0.01% in this case), significantly improved chromatographic performance. Good peak shapes and resolution of EtG and EtS from the complex matrix interferences in this particularly problematic urine sample were obtained, allowing for confident quantitation.
Note that blank urine samples prepared by protein precipitation with acetonitrile and subsequent aqueous dilution will take longer to condition a new column compared to blank urine samples prepared using a dilute-and-shoot method.
Figure 8: Impact of Sample Preparation on Retention and Peak Shape.
| Peaks | Conc. (ng/mL) | Precursor Ion | Product Ion | Product Ion | |
|---|---|---|---|---|---|
| 1. | Ethyl-β-D-glucuronide-d5 (EtG-d5) | 200 | 226.2 | 85.0 | - |
| 2. | Ethyl-β-D-glucuronide (EtG) | - | 221.2 | 75.1 | 85.1 |
| 3. | Ethyl sulfate-d5 (EtS-d5) | 50 | 130.1 | 98.0 | - |
| 4. | Ethyl sulfate (EtS) | - | 125.1 | 97.1 | 80.0 |
| Column | Raptor EtG/EtS (cat.# 9325A12) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 100 mm x 2.1 mm ID | ||||||||||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||||||||||
| Pore Size: | 90 Å | ||||||||||||||||||||
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 μm frit (cat.# 25809) | ||||||||||||||||||||
| Temp.: | 35 °C | ||||||||||||||||||||
| Standard/Sample | Ethyl- | ||||||||||||||||||||
| Ethyl sulfate-d5 sodium salt (EtS-d5) (cat.# 34104) | |||||||||||||||||||||
| Diluent: | 0.01% Formic acid in water | ||||||||||||||||||||
| Conc.: | Human urine sample containing endogenous EtG and EtS | ||||||||||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||||||||||
| Mobile Phase | |||||||||||||||||||||
| A: | 0.01% Formic acid in water | ||||||||||||||||||||
| B: | 0.1% Formic acid in acetonitrile | ||||||||||||||||||||
|
| Detector | MS/MS |
|---|---|
| Ion Source: | Electrospray |
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | HPLC |
| Sample Preparation | Dilute and Shoot: A 50 μL aliquot was taken from a urine sample containing endogenous EtG and EtS and diluted 20 times with 950 µL of a working internal standard (200 ng/mL EtG-d5 and 50 ng/mL EtS-d5 in 0.01% formic acid in water). The sample was vortexed at 3000 rpm for 10 seconds to mix. The sample was then centrifuged at 4300 rpm for 10 minutes at 10 °C and the supernatant was injected. Acetonitrile Precipitation: A 50 μL aliquot was taken from a urine sample containing endogenous EtG and EtS. 10 μL of internal standard (20 μg EtG-d5 and 5 μg EtS-d5) and 150 µL of acetonitrile were added to the aliquot. The sample was vortexed at 3000 rpm for 10 seconds to mix and centrifuged at 4300 rpm for 10 minutes at 10 °C. After centrifugation, 100 µL of the supernatant was aliquoted and injected for the 4x level (without further dilution) or diluted again with 400 µL (20x) or 900 µL (40x) of 0.01% formic acid in water, respectively. The samples were then vortexed at 3000 rpm for 10 seconds and injected. |
Increased Method Versatility through Combined Strategies
All of the strategies discussed here can be combined to develop an LC-MS/MS method that will provide greater versatility, which is especially important for laboratories that process very high volumes of highly variable samples.
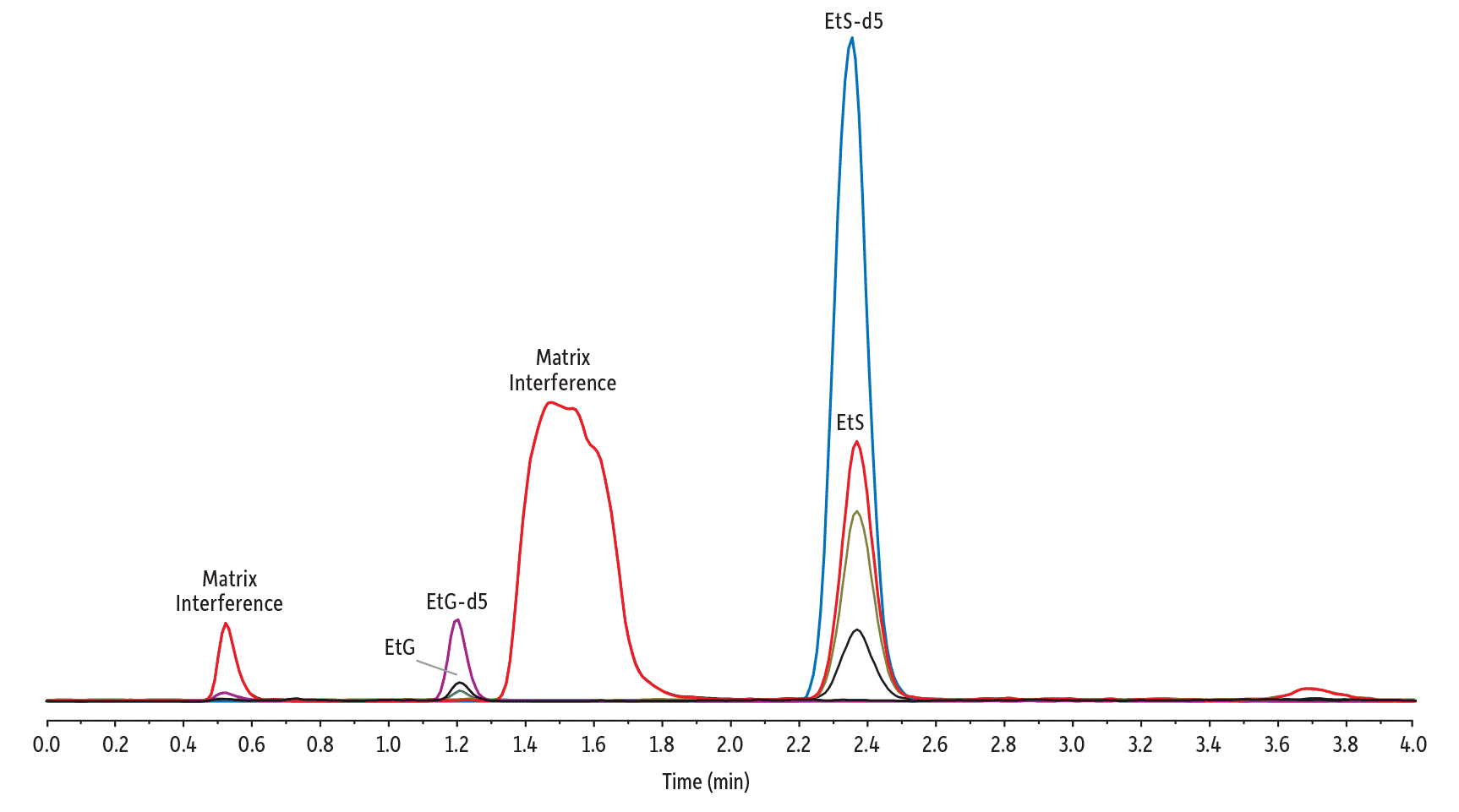
Figure 9 is a demonstration of these techniques being integrated and used for a sample that proved problematic for dilute-and-shoot sample preparation alone. The addition of protein precipitation/dilution and the 0.5 minute extension of the gradient program do increase the sample preparation and analysis time, but for labs responsible for analyzing high volumes of samples every day, the added insurance may well be worth it. In summary, a combined strategy of optimized instrument setup, sample preparation, and method conditions provides excellent sensitivity and resolution from matrix interferences for the analysis of EtG and EtS in urine.
Figure 9: Good sensitivity and separation were achieved for the analysis of EtG and EtS in urine on a Raptor EtG/EtS column using a combined analytical strategy.
| Peaks | tR (min) | Conc. (ng/mL) | Precursor Ion | Product Ion | Product Ion | |
|---|---|---|---|---|---|---|
| 1. | Ethyl-β-D-glucuronide-d5 (EtG-d5) | 1.21 | 200 | 226.2 | 85.0 | - |
| 2. | Ethyl-β-D-glucuronide (EtG) | 1.23 | 500 | 221.2 | 75.1 | 85.1 |
| 3. | Ethyl sulfate-d5 (EtS-d5) | 2.36 | 50 | 130.1 | 98.0 | - |
| 4. | Ethyl sulfate (EtS) | 2.38 | 500 | 125.1 | 97.1 | 80.0 |
| Column | Raptor EtG/EtS (cat.# 9325A12) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dimensions: | 100 mm x 2.1 mm ID | ||||||||||||||||||||
| Particle Size: | 2.7 µm | ||||||||||||||||||||
| Pore Size: | 90 Å | ||||||||||||||||||||
| Guard Column: | UltraShield UHPLC precolumn filter, 0.2 μm frit (cat.# 25809) | ||||||||||||||||||||
| Temp.: | 35 °C | ||||||||||||||||||||
| Standard/Sample | Ethyl- | ||||||||||||||||||||
| Ethyl- | |||||||||||||||||||||
| Ethyl sulfate-d5 sodium salt (EtS-d5) (cat.# 34104) | |||||||||||||||||||||
| Ethyl sulfate sodium salt (EtS) (cat.# 34103) | |||||||||||||||||||||
| Diluent: | 0.01% Formic acid in water | ||||||||||||||||||||
| Conc.: | 500 ng/mL (40x acetonitrile precipitation sample prep) | ||||||||||||||||||||
| Inj. Vol.: | 10 µL | ||||||||||||||||||||
| Mobile Phase | |||||||||||||||||||||
| A: | 0.01% Formic acid in water | ||||||||||||||||||||
| B: | 0.1% Formic acid in acetonitrile | ||||||||||||||||||||
|
| Detector | MS/MS |
|---|---|
| Ion Source: | Electrospray |
| Ion Mode: | ESI- |
| Mode: | MRM |
| Instrument | HPLC |
| Sample Preparation | A 500 ng/mL standard was prepared in urine. A 50 μL aliquot was mixed with 10 μL of internal standard (20 μg/mL EtG-d5 and 5 μg/mL EtS-d5 in water) and 150 µL of acetonitrile by vortexing at 3000 rpm for 10 seconds and centrifuged at 4300 rpm for 10 minutes at 10 °C. After centrifugation, 100 µL of the supernatant was diluted with 900 µL (40x dilution) of 0.01% formic acid in water. The sample was then vortexed at 3000 rpm for 10 seconds and injected for LC-MS/MS analysis. |